
الگوریتم AlphaFold: راهکاری برای چالش 50 سالهی زیستشناسی
 تیم تحریریه
تیم تحریریه- ۴ مرداد ۱۴۰۰
پروتئینها عنصری ضروری برای حیات و زیربنای تمامی کارکردهای آن هستند. پروتئینها مولکولهای بزرگ و پیچیدهای هستند که از زنجیرههای آمینواسید ساخته شدهاند. کار این مولکولها عمدتاً توسط ساختار سهبُعدی خاص آنها تعیین میشود. شناخت شکل تاخوردگی مولکول پروتئین به عنوان «مسئلهی تاخوردگی پروتئین Protein folding problem» شناخته میشود؛ نزدیک 50 سال است که این مسئله از چالشهای عظیم حوزهی زیستشناسی به شمار میرود. ما در جدیدترین نسخهی سیستم هوش مصنوعی خود به نام الگوریتم AlphaFold به پیشرفت علمی بزرگی دست یافتیم. بنیانگذاران انجمن CASP (ارزیابی بنیادین پیشبینی ساختار پروتئین Critical Assessment of protein Structure Prediction) این سیستم را راهکاری برای این چالش بزرگ دانستهاند. این کشف عظیم حاکی از تأثیرگذاری هوش مصنوعی بر روی کشفیات علمی و نشاندهندهی ظرفیت این حوزه در تسریع چشمگیر علوم پایهای است که جهان ما را شکل داده و معنا بخشیدهاند.
شکل یک پروتئین با کارکرد (وظیفهی) آن ارتباطی نزدیک دارد و زمانی که بتوانیم ساختار پروتئین را پیشبینی کنیم، درکی جامع از چیستی و چگونگی کار آن به دست میآوریم. بسیاری از چالشهای بزرگ دنیا، همچون یافتن درمان برای بیماریها یا کشف آنزیمهایی که ضایعات صنعتی را کاهش میدهند، رابطهای تنگاتنگ با پروتئینها و وظایف آنها دارند.
پروفسور جان مالت، یکی از بنیانگذاران و رئیس هیأت مدیرهی CASP دانشگاه مریلند، میگوید: « نزدیک 50 سال این سؤال ذهن دانشمندان را به خود مشغول کرده است که پروتئینها چگونه تا میخورند؟ به عنوان کسی که مدت زمان زیادی را صرف این مسئله کردهام، یافتن راهکاری توسط شرکت DeepMind، لحظهای به یاد ماندنی به شمار میرود. »
این کار حاصل سالها پژوهش گستردهی علمی در حوزهی مطالعه و تعیین ساختار پروتیئنها با استفاده از تکنیکهای تجربی گوناگون، همچونرزونانس مغناطیسی هستهای Nuclear Magnetic Resonance (MNR) و بلورنگاری اشعهایکس X-Ray Crystallography، بوده است. تکنیکها و روشهای جدیدتر مثل میکروسکوپ الکترونی کرایو Cryo-electron microscopy بر عملیات آزمون و خطا تکیه دارند که برای تعیین هر ساختار نیازمند سالها زحمت و کار سخت و تجهیزات تخصصی چندین میلیون دلاری هستند.
مسئلهی تاخوردگی پروتئین
کریسشن انفینزن در سخنرانی معروف خود پس از دریافت جایزهی نوبل شیمی در سال 1972 بیان کرد که «به صورت نظری، توالی آمینواسیدهای یک مولکول پروتئین، ساختار پروتئین را به صورت کامل تعیین میکند.» این فرضیه نقطهی آغاز تلاشی 50 ساله برای پیشبینی ساختار سهبُعدی پروتئینها از طریق محاسبات و تنها با استفاده از توالی یکبُعدی آمینواسیدی، به جای روشهای تجربی زمانبر و گرانقیمت بود. با این حال یک چالش بزرگ وجود داشت: به صورت نظری، تعداد حالتهای ممکن برای تاشدگی یک پروتئین قبل از قرارگیری در ساختار سهبُعدی نهایی، بیشمار است. سایرس لوینتال Cyrus Levinthal در سال 1960 به این نکته اشاره کرد که شمردن همهی ترکیبات احتمالی یک پروتئین ساده به زمانی بیشتر از عمر هستی نیاز خواهد داشت؛ طبق برآورد وی ترکیب احتمالی برای یک پروتئین ساده وجود دارد. این درحالی است که در طبیعت، پروتئینها به صورت خودبهخودی، بعضاً طی یکهزارم ثانیه تا میخورند. از این دوگانگی گاهی به عنوان پارادوکس لوینتال Levinthal’s paradox یاد میشود.
ویدئو توضیح تاشدگی پروتئینها
یافتههای ارزیابی CASP14
در سال 1994، پروفسور جان مالت و پروفسور کریستوف فیدلیس انجمن CASP را بنیانگزاری کردند؛ این انجمن یک مرکز ارزیابی است که مأموریتش کاتالیز پژوهشها، نظارت بر پیشرفت و احراز آخرین کشفیات در مورد پیشبینی ساختار پروتئین است. CASP را میتوان یک استاندارد طلایی برای ارزیابی تکنیکهای پیشبینی و همچنین یک انجمن جهانی منحصر به فرد دانست که بر اساس تلاشی مشترک بنا شده است. CASP آخرین و جدیدترین ساختارهای پروتئین (برخی از این ساختارها در زمان ارزیابی هنوز در انتظار تأیید بودند) را که به صورت تجربی تعیین شدهاند، انتخاب میکند تا تیمها برای آزمون روشهای پیشبینی ساختار که طراحی کردهاند، به کار ببرند (مقایسهی ساختارهایی که توسط مدلهای هوش مصنوعی پیشبینی میشود با ساختارهایی که در طرحهای آزمایشی تعیین میشود). شرکتکنندگان باید ساختار پروتئینها را به صورت کورکورانه پیشبینی کنند؛ سپس این پیشبینیها با دادههای آزمایشی حقیقتپایه مقایسه میشوند. ما این کشف عظیم را وامدار مؤسسان و همگی اعضای CASP و آزمایشگرانی هستیم که ساختارهایشان چنین ارزیابی دقیقی را میسر کردهاند.
ویدئو سیستم هوش مصنوعی آلفافولد: پیشرفتی غیرمنتظره برای علم
معیار اصلی که CASP برای اندازهگیری میزان دقت پیشبینیها به کار میبرد GDT یا آزمون فاصلهی کلی است که میتواند بین 0 تا 100 باشد. به بیان ساده، GDT را میتوان درصد باقیماندههای آمینواسید (اجزای زنجیرهی پروتئین) در یک فاصلهی آستانهای از موقعیت درست دانست. طبق سخنان پرفسور مالت، ساختاری که نمره حدود 90 در GDT به دست آورد را میتوان به صورت غیررسمی قابل مقایسه با نتایج روشهای تجربی درنظر گرفت.
یافتههای چهاردهمین ارزیابی CASP نشان میدهند میانهی نمراتی که آخرین نسخهی سیستم AlphaFold در GDT در بین اهداف (نمونههای پیشبینی) گوناگون به دست آورده برابر با 92.4 است. این بدین معنی است که میانگین خطای پیشبینیهای ما (RMSD) حدود 1.6 آنگستروم Angstroms ، یعنی چیزی حدود عرض یک اتم (یا 0.1 یک نانومتر) میباشد. AlphaFold حتی برای سختترین پروتئینهای هدف (آنهایی که در چالشبرانگیزترین دستهی مدلسازی آزاد Free-modelling قرار میگیرند) نیز میانهی نمرات 87.0 را به دست میآورد(دادهها در این لینک موجود هستند).
میانه میزان دقت مدلسازی آزاد


با این نتایج امیدوارکننده میتوان پیشبینی ساختار از طریق محاسبات را یکی از ابزارهای اصلی در پژوهشهای علمی برای زیستشناسان درنظر گرفت. یکی از مزایای روشهای ما مربوط به دستههای مهمی از پروتئینها (مثل پروتئینهای غشاء) میشود که متبلورسازی و در نتیجه تعیین ساختارشان به صورت تجربی کار دشوار و چالشبرانگیزی است.
پروفسور ونکی راماکریشنان Venki Ramakrishnan، از برندگان نوبل و رئیس Royal Society، میگوید: «این محاسبات پیشرفتی عظیم در مسئلهی تاشدگی پروتئین، این چالش بزرگ و قدیمی زیستشناسی، به شمار میروند. تحولاتی که این روش در تحقیقات زیستشناسی به وجود خواهد آورد، دیدنی خواهد بود.»
رویکرد ما در مقابل مسئلهی تاشدگی پروتئین
در سال 2018، ما با نسخهی اولیهی الگوریتم AlphaFold وارد رقابتهای CASP13 شدیم و به بالاترین میزان دقت در میان شرکتکنندگان دست یافتیم. بعد از آن در مورد روشهای ارزیابیشده در CASP13 مقالهای در ژورنال Nature منتشر کردیم که کد را هم به همراه داشت؛ این مقاله الهامبخش کارهای پژوهشی و کاربردی متنباز دیگری بود که توسط انجمن طراحی شدند. معماریهای جدید یادگیری عمیقی که طراحی کردهایم، محرکهی تغییر در روشهای به کاررفته در CASP14 بودهاند و ما را قادر میسازند به دقتی بیهمتا دست پیدا کنیم. این روشها از حوزههای زیستشناسی، فیزیک و یادگیری ماشینی، و البته کارهای فراوانی که طی نیمقرن گذشته توسط دانشمندان در زمینهی تاشدگی پروتئین انجام شده، الهام گرفتهاند.
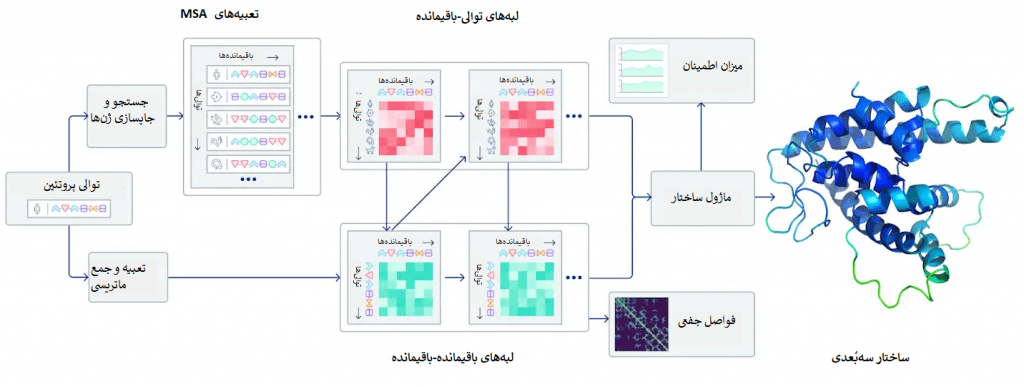
یک پروتئین تاشده را میتوان به عنوان یک «نمودار فضایی Spatial graph» در نظر گرفت که در آن باقیماندهها گرهها هستند و لبهها باقیماندهها را در فواصل نزدیک به هم متصل میکنند. این نمودار برای درک تعاملات فیزیکی درون پروتئینها و تاریخچهی تحولی آنها مهم است. در آخرین نسخهی الگوریتم AlphaFold که در CASP14 استفاده شد، یک سیستم شبکهی عصبی توجهمحور ساختیم که به صورت نقطهبهنقطه آموزش دیده و سعی میکند ساختار آن نمودار را تفسیر کند و در عین حال در مورد نمودار ضمنی (که ساختار است) استدلال نماید. این سیستم از توالیهای مرتبط با روند تحولی، MSA (همترازسازی چند توالی Multiple sequence alignment ) و یک بازنمایی از جفتباقیماندههای آمینواسیدها برای پالایش نمودار استفاده میکند.
سیستم با تکرار این فرآیند، پیشبینیهایی دقیق در مورد ساختار فیزیکی زیربنایی پروتئین تولید میکند و میتواند ساختار پروتئینها را به طور بسیار دقیق و تنها طی چندروز تعیین کند. علاوه بر اینها، AlphaFold قادر است با استفاده از یک معیار اطمینان درونی پیشبینی کند کدام قسمتها از ساختار پیشبینیشده قابل اتکا (معتبر) هستند.
ما این سیستم را با استفاده از یک دیتاست عمومی شامل حدود 170000 ساختار پروتئین که از بانک دادههای پروتئینی Protein data bank و پایگاههای دادهای بزرگ (که توالیهای پروتئینی از ساختارهای ناشناخته را در بردارند) گرفته شدند، آموزش دادیم. این سیستم تقریباً از شانزده TPUv3 (TPUv3 128هستهای که حدوداً برابر با 100-200 GPU است) استفاده میکند و اجرای آن بیشتر از چند هفته طول میکشد، که در مقایسه با حجم محاسباتی موردنیاز در بیشتر مدلهای بزرگ موجود در یادگیری ماشینی، حجم محاسباتی نسبتاً خوبی به شمار میرود. همچون سیستمی که در CASP13 استفاده کردیم، این بار هم در حال نگارش یک مقاله برای یک ژورنال داوری شده مناسب هستیم.
تأثیرات این مدل بر دنیای واقعی
ده سال پیش که شرکت DeepMind تأسیس شد، امیدوار بودیم روزی فرا برسد که کشفیات حوزهی هوش مصنوعی بتواند زمینهی حل مسائل پیشرفتهی علوم پایه را فراهم کند. حال، بعد از 4 سال تلاش برای ساخت AlphaFold، میتوانیم شاهد محقق شدن این هدف و کمک به حوزههایی همچون ساخت دارو و حفظ محیط زیست باشیم.
پروفسور آندری لوپاس Andrei Lupas، مدیر مؤسسهی زیستشناسی رشدی مکس پلانک Max Planck Institute for Developmental Biology و یکی از ارزیابان CASP معتقد است: « مدلهای بسیار دقیق AlphaFold ما را قادر ساختهاند ساختار یک پروتئین را که نزدیک به یک دهه نامعلوم بوده، پیشبینی کرده و نهایتاً درک کنیم که سیگنالها چگونه در سطح غشاءهای سلولی منتقل میشوند. »
ما در مورد تأثیرات احتمالی AlphaFold روی پژوهشهای زیستشناسی و دنیای واقعی، خوشبین هستیم و مشتاقیم با همکاری دیگران ظرفیتهای آتی آن را کشف کنیم. همزمان با کار روی مقالهی مذکور، به دنبال این هستیم که امکان دسترسی گسترده را به این سیستم مهیا نماییم.
در حال حاضر با همکاری چندین گروه از متخصصان، در حال مطالعهی کابرد پیشبینی ساختار پروتئینها در شناخت بیماریهای خاص هستیم، مثلاً اینکه به ما کمک میکند پروتئینهایی که کژکارکردی داشتهاند را تشخیص داده و در مورد نحوهی تعامل آنها استدلال کنیم. علم به این نکات ما را قادر میسازد در طراحی دارو دقیقتر عمل کنیم و بدین ترتیب به مکملی برای روشهای تجربی موجود دست یابیم که به سرعت به درمانهای امیدوارکننده میرسد.
به گفتهی دکتر آرتور دیلوینسون Arthur D. Levinson، مؤسس و مدیرعامل Calico، رئیس هیأت مدیره و مدیرعامل سابق Genetech، سیستم AlphaFold پیشرفتی تاریخی و منحصر به فرد است که ساختار پروتئینها را با سرعت و دقت عالی پیشبینی میکند. این گام رو به جلو به ما نشان میدهد روشهای محاسباتی آمادهی تغییر تحقیقات زیستشناسی هستند و چشمانداز امیدوارکنندهای برای فرآیند کشف دارو پیش رو قرار میدهند.
برخی از نشانههای مشاهدهشده نیز ما را بر این باور داشته که پیشبینی ساختار پروتئین میتواند در مدیریت پاندمیکهای آینده مفید باشد و به عنوان یکی از ابزارهای متعددی که توسط جامعهی دانشمندان ساخته شده، مورد استفاده قرار گیرد. اوایل سال جاری چندین ساختار پروتئین (از جمله ORF3a) از ویروس SARS-CoV-2 پیشبینی کردیم که پیش از آن ناشناخته بودند. در CASP14 نیز یک ساختار پروتئینی دیگر (ORF8) از ویروس کرونا پیشبینی کردیم. اکنون کارهای بسیار سریع آزمایشگران ساختارهای ORF3a و ORF8 را تأیید کرده است. ما توانستیم با وجود ذات چالشبرانگیز کار و در دست داشتن توالیهای بسیار معدود، (در مقایسه با ساختارهایی که به صورت تجربی تعیین شده بودند) به سطح بالایی از دقت در هردو پیشبینی خود دست یابیم.
امیدواریم این تکنیکها، علاوه بر شتاببخشی به درکی که از بیماریهای شناختهشده داریم، در جستجوی صدها میلیون پروتئینی که هنوز مدلی برایشان نداریم (و حوزهای وسیع از زیستشناسی را تشکیل میدهند) نیز به ما کمک کنند. از آنجایی که DNA توالی آمینواسیدها را (که ساختار پروتئینها را تشکیل میدهند) مشخص میکند، تحولات حوزهی ژنومیک امکان خواندن توالی پروتئینهای دنیای واقعی در مقیاسهای بزرگ (180 میلیون توالی پروتئینی در پایگاه داده جهانی پروتئینی Universal Protein Database یا UniProt) را فراهم آورده است. از سوی دیگر با توجه به کار تجربی موردنیاز برای تشخیص ساختار از روی توالیها، تنها حدود 170000 ساختار پروتئینی در بانک داده پروتئین Protein Data Bank (PDB) قرار دارد. ممکن است برخی از این پروتئینهای تعییننشده کارکردی جدید و جذاب داشته باشند و AlphaFold، همچون تلسکوپی که به ما کمک میکند نگاهی عمیقتر به جهان ناشناختهها داشته باشیم، ما را در یافتن این کارکردها و ویژگیها یاری میکند.
کشف قابلیتهای جدید
الگوریتم AlphaFold یکی از چشمگیرترین پیشرفتهایی است که تاکنون حاصل شده و همچون هر پژوهش علمی دیگری، هنوز سؤالاتی بدون پاسخ دارد. همهی ساختارهایی که با این مدل پیشبینی میکنیم عالی نیستند. هنوز چیزهای زیادی برای یادگیری وجود دارند، از جمله اینکه پروتئینها چطور ترکیبات را شکل میدهند، تعاملشان با DNA، RNA و مولکولهای کوچک چگونه است و چطور میتوان موقعیت مکانی همهی زنجیرههای آمینواسیدی جانبی را تشخیص داد. امیدواریم با همکاری سایرین این را هم بیاموزیم که چگونه میتوان برای طراحی داروهای جدید، روشهایی نوین برای مدیریت محیطزیست و موارد مشابه، به بهترین نحو ممکن از کشفیات علمی بهره برد.
برای همهی افرادی که روی روشهای محاسباتی و یادگیری ماشینی در علم کار میکنند، سیستمهایی همچون الگوریتم AlphaFold نمایانگر پتانسیل خارقالعادهی هوش مصنوعی به عنوان ابزاری برای کمک به کشفیات بنیادین است. همانطور که آنفینسن 50 سال قبل چالشی را مطرح کرد که بسیار فراتر از محدودهی علم آن زمان بود، در حال حاضر نیز جنبههای فراوانی از جهان اطراف ما وجود دارد که ناشناخته باقی مانده است. پیشرفتی که امروزه به آن دست یافتیم به ما اطمینان بیشتری میبخشد که هوش مصنوعی به یکی از مفیدترین ابزارها برای پیشبرد دانش تبدیل خواهد شد و باید آمادهی سالها تلاش سخت و کشفیات جدید باشیم.
جدیدترین اخبار هوش مصنوعی ایران و جهان را با هوشیو دنبال کنید.





